by Mariya Dimitrova, Jorrit Poelen, Georgi Zhelezov, Teodor Georgiev, Lyubomir Penev

Tables published in scholarly literature are a rich source of primary biodiversity data. They are often used for communicating species occurrence data, morphological characteristics of specimens, links of species or specimens to particular genes, ecology data and biotic interactions between species, etc. Tables provide a structured format for sharing numerous facts about biodiversity in a concise and clear way.
Together with the rest of the article narrative, Pensoft publishes all tables in the semi-structured eXtensible Markup Language (XML) format. Tables are semantically enhanced with annotated taxonomic names, coordinates, localities and other fields from the Darwin Core Standard.
Inspired by the potential use of semantically-enhanced tables for text and data mining, Pensoft and Global Biotic Interactions (GloBI) developed a workflow for extracting and indexing biotic interactions from tables published in scholarly literature. GloBI is an open infrastructure enabling the discovery and sharing of species interaction data. GloBI ingests and accumulates individual datasets containing biotic interactions and standardises them by mapping them to community-accepted ontologies, vocabularies and taxonomies. Data integrated by GloBI is accessible through an application programming interface (API) and as archives in different formats (e.g. n-quads). GloBI has indexed millions of species interactions from hundreds of existing datasets spanning over a hundred thousand taxa.
The workflow
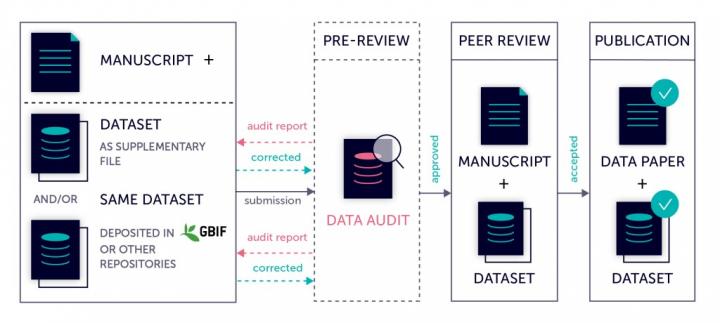
First, all tables extracted from Pensoft publications and stored in the OpenBiodiv triple store were automatically retrieved (Step 1 in Fig. 1). There were 6993 tables from 21 different journals. To identify only the tables containing biotic interactions, we used an ontology annotator, currently developed by Pensoft using terms from the OBO Relation Ontology (RO). The Pensoft Annotator analyses free text and finds words and phrases matching ontology term labels.
We used the RO to create a custom ontology, or list of terms, describing different biotic interactions (e.g. ‘host of’, ‘parasite of’, ‘pollinates’) (Step 2 in Fig. 1).. We used all subproperties of the RO term labeled ‘biotically interacts with’ and expanded the list of terms with additional word spellings and variations (e.g. ‘hostof’, ‘host’) which were added to the custom ontology as synonyms of already existing terms using the property oboInOwl:hasExactSynonym.
This custom ontology was used to perform annotation of all tables via the Pensoft Annotator (Step 3 in Fig. 1). Tables were split into rows and columns and accompanying table metadata (captions). Each of these elements was then processed through the Pensoft Annotator and if a match from the custom ontology was found, the resulting annotation was written to a MongoDB database, together with the article metadata. The original table in XML format, containing marked-up taxa, was also stored in the records.
Thus, we detected 233 tables which contain biotic interactions, constituting about 3.4% of all examined tables. The scripts used for parsing the tables and annotating them, together with the custom ontology, are open source and available on GitHub. The database records were exported as json to a GitHub repository, from where they could be accessed by GloBI.
GloBI processed the tables further, involving the generation of a table citation from the article metadata and the extraction of interactions between species from the table rows (Step 4 in Fig. 1). Table citations were generated by querying the OpenBiodiv database with the DOI of the article containing each table to obtain the author list, article title, journal name and publication year. The extraction of table contents was not a straightforward process because tables do not follow a single schema and can contain both merged rows and columns (signified using the ‘rowspan’ and ‘colspan’ attributes in the XML). GloBI were able to index such tables by duplicating rows and columns where needed to be able to extract the biotic interactions within them. Taxonomic name markup allowed GloBI to identify the taxonomic names of species participating in the interactions. However, the underlying interaction could not be established for each table without introducing false positives due to the complicated table structures which do not specify the directionality of the interaction. Hence, for now, interactions are only of the type ‘biotically interacts with’ (Fig. 2) because it is a bi-directional one (e.g. ‘Species A interacts with Species B’ is equivalent to ‘Species B interacts with Species A’).
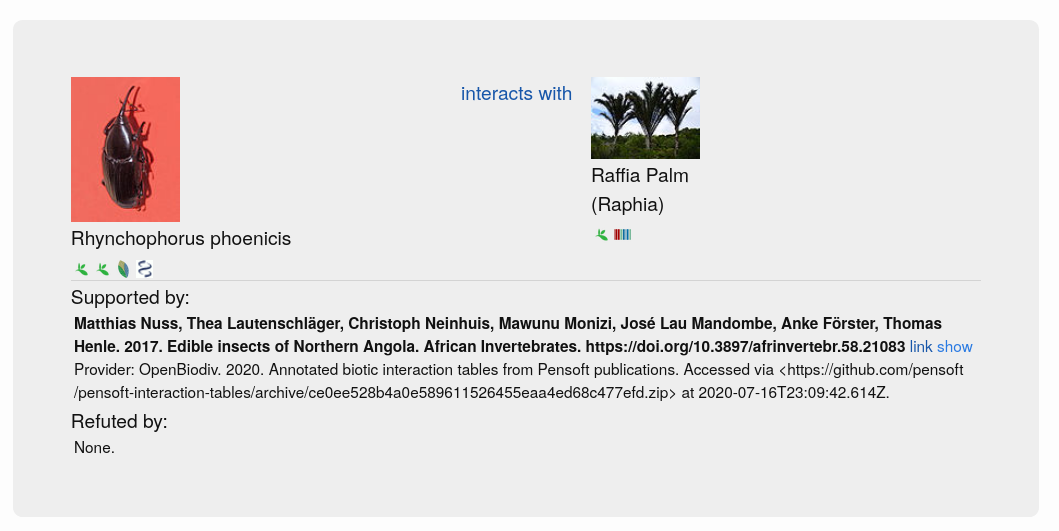
Examples of species interactions provided by OpenBiodiv and indexed by GloBI are available on GloBI’s website.
In the future we plan to expand the capacity of the workflow to recognise interaction types in more detail. This could be implemented by applying part of speech tagging to establish the subject and object of an interaction.
In addition to being accessible via an API and as archives, biotic interactions indexed by GloBI are available as Linked Open Data and can be accessed via a SPARQL endpoint. Hence, we plan on creating a user-friendly service for federated querying of GloBI and OpenBiodiv biodiversity data.
This collaborative project is an example of the benefits of open and FAIR data, enabling the enhancement of biodiversity data through the integration between Pensoft and GloBI. Transformation of knowledge contained in existing scholarly works into giant, searchable knowledge graphs increases the visibility and attributed re-use of scientific publications.
Tables published in scholarly literature are a rich source of primary biodiversity data. They are often used for communicating species occurrence data, morphological characteristics of specimens, links of species or specimens to particular genes, ecology data and biotic interactions between species etc. Tables provide a structured format for sharing numerous facts about biodiversity in a concise and clear way.
Together with the rest of the article narrative, Pensoft publishes all tables in the semi-structured eXtensible Markup Language (XML) format. Tables are semantically enhanced with annotated taxonomic names, coordinates, localities and other fields from the Darwin Core Standard.
Inspired by the potential use of semantically-enhanced tables for text and data mining, Pensoft and Global Biotic Interactions (GloBI) developed a workflow for extracting and indexing biotic interactions from tables published in scholarly literature. GloBI is an open infrastructure enabling the discovery and sharing of species interaction data. GloBI ingests and accumulates individual datasets containing biotic interactions and standardises them by mapping them to community-accepted ontologies, vocabularies and taxonomies. Data integrated by GloBI is accessible through an application programming interface (API) and as archives in different formats (e.g. n-quads). GloBI has indexed millions of species interactions from hundreds of existing datasets spanning over a hundred thousand taxa.
The workflow
First, all tables extracted from Pensoft publications and stored in the OpenBiodiv triple store were automatically retrieved (Step 1 in Fig. 1). There were 6,993 tables from 21 different journals. To identify only the tables containing biotic interactions, we used an ontology annotator, currently developed by Pensoft using terms from the OBO Relation Ontology (RO). The Pensoft Annotator analyses free text and finds words and phrases matching ontology term labels.
We used the RO to create a custom ontology, or list of terms, describing different biotic interactions (e.g. ‘host of’, ‘parasite of’, ‘pollinates’) (Step 1 in Fig. 1). We used all subproperties of the RO term labeled ‘biotically interacts with’ and expanded the list of terms with additional word spellings and variations (e.g. ‘hostof’, ‘host’) which were added to the custom ontology as synonyms of already existing terms using the property oboInOwl:hasExactSynonym.
This custom ontology was used to perform annotation of all tables via the Pensoft Annotator (Step 3 in Fig. 1). Tables were split into rows and columns and accompanying table metadata (captions). Each of these elements was then processed through the Pensoft Annotator and if a match from the custom ontology was found, the resulting annotation was written to a MongoDB database, together with the article metadata. The original table in XML format, containing marked-up taxa, was also stored in the records.
Thus, we detected 233 tables which contain biotic interactions, constituting about 3.4% of all examined tables. The scripts used for parsing the tables and annotating them, together with the custom ontology, are open source and available on GitHub. The database records were exported as JSON to a GitHub repository, from where they could be accessed by GloBI.
GloBI processed the tables further, involving the generation of a table citation from the article metadata and the extraction of interactions between species from the table rows (Step 4 in Fig. 1). Table citations were generated by querying the OpenBiodiv database with the DOI of the article containing each table to obtain the author list, article title, journal name and publication year. The extraction of table contents was not a straightforward process because tables do not follow a single schema and can contain both merged rows and columns (signified using the ‘rowspan’ and ‘colspan’ attributes in the XML). GloBI were able to index such tables by duplicating rows and columns where needed to be able to extract the biotic interactions within them. Taxonomic name markup allowed GloBI to identify the taxonomic names of species participating in the interactions. However, the underlying interaction could not be established for each table without introducing false positives due to the complicated table structures which do not specify the directionality of the interaction. Hence, for now, interactions are only of the type ‘biotically interacts with’ because it is a bi-directional one (e.g. ‘Species A interacts with Species B’ is equivalent to ‘Species B interacts with Species A’).
In the future, we plan to expand the capacity of the workflow to recognise interaction types in more detail. This could be implemented by applying part of speech tagging to establish the subject and object of an interaction.
In addition to being accessible via an API and as archives, biotic interactions indexed by GloBI are available as Linked Open Data and can be accessed via a SPARQL endpoint. Hence, we plan on creating a user-friendly service for federated querying of GloBI and OpenBiodiv biodiversity data.
This collaborative project is an example of the benefits of open and FAIR data, enabling the enhancement of biodiversity data through the integration between Pensoft and GloBI. Transformation of knowledge contained in existing scholarly works into giant, searchable knowledge graphs increases the visibility and attributed re-use of scientific publications.
References
Jorrit H. Poelen, James D. Simons and Chris J. Mungall. (2014). Global Biotic Interactions: An open infrastructure to share and analyze species-interaction datasets. Ecological Informatics. https://doi.org/10.1016/j.ecoinf.2014.08.005.
Additional Information
The work has been partially supported by the International Training Network (ITN) IGNITE funded by the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 764840.