October and November 2023 were active months for the Pensoft team, who represented the publisher’s journals and projects at conferences in Europe, North America, South America, Oceania and Asia.
Let’s take a look back at all the events of the past two months.
The Biodiversity Information Standards Conference 2023
The Biodiversity Information Standards (TDWG) Conference, held from October 9-13 in Tasmania, Australia, brought together experts and stakeholders from the global biodiversity research community.

The annual gathering is a crucial platform for sharing insights, innovations, and knowledge related to biodiversity data standards and practices. Key figures from Pensoft took part in the event, presenting new ways to improve the management, accessibility, and usability of biodiversity data.
Prof. Lyubomir Penev, founder and Chief Executive Officer of Pensoft, gave two talks that highlighted the importance of data publishing. His presentation on “The Biodiversity Knowledge Hub (BKH): A Crosspoint and Knowledge Broker for FAIR and Linked Biodiversity Data” underscored the significance of FAIR (Findable, Accessible, Interoperable, and Reusable) data standards. BKH is the major output from the Horizon 2020 project BiCIKL (Biodiversity Community Integrated Knowledge Library) dedicated to linked and FAIR data in biodiversity, and coordinated by Pensoft.

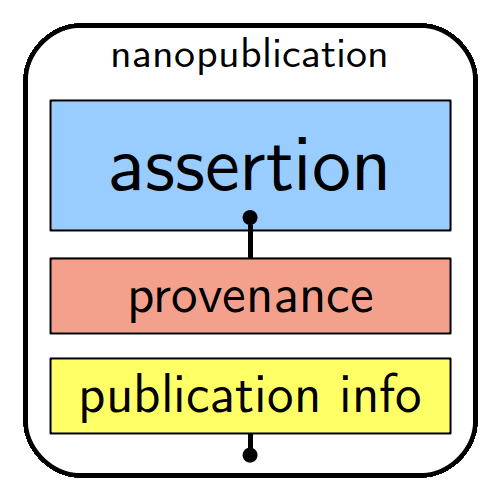
He also introduced the Nanopublications for Biodiversity workflow and format: a promising new tool developed by Knowledge Pixels and Pensoft to communicate key scientific statements in a way that is human-readable, machine-actionable, and in line with FAIR principles. Earlier this year, Biodiversity Data Journal integrated nanopublications into its workflow to allow authors to share their findings even more efficiently.
Chief Technology Officer of Pensoft Teodor Georgiev contributed to the conference by presenting “OpenBiodiv for Users: Applications and Approaches to Explore a Biodiversity Knowledge Graph.” His session highlighted the innovative approaches being taken to explore and leverage a biodiversity knowledge graph, showcasing the importance of technology in advancing biodiversity research.

Many authors and editors at Biodiversity Data Journal also spoke at the TDWG conference, including Vince Smith, the journal’s editor-in-chief, who is Head of Digital, Data, and Informatics at the Natural History Museum. He delivered insightful presentations on digitising natural science collections and utilising AI for insect collections.
GEO BON Global Conference 2023
GEO BON’s Global Conference on Biodiversity and Monitoring took place from 10-13 October 2023 in Montreal, Canada.

The theme of the conference was “Monitoring Biodiversity for Action” and there was particular emphasis on the development of best practices and new technologies for biodiversity observations and monitoring to support transformative policy and conservation action.
Metabarcoding & Metagenomics’ editor-in-chief, Florian Leese, was one of the organisers of the “Standardized eDNA-Based Biodiversity Monitoring to Inform Environmental Stewardship Programs” session. Furthermore, the journal was represented at Pensoft’s exhibition booth, where conference participants were able to discuss metabarcoding and metagenomics research.
Following the conference, Metabarcoding & Metagenomics announced a new special issue titled “Towards Standardized Molecular Biodiversity Monitoring.” The special issue is accepting submissions until 15th March 2024.
Asian Mycological Congress 2023
The Asian Mycological Congress welcomed researchers from around the world to Busan, Republic of Korea, for an exploration of all things fungi from 10-13 October.

Titled “Fungal World and Its Bioexploitation – in all areas of basic and applied mycology,” the conference covered a range of topics related to all theoretical and practical aspects of mycology. There was a particular emphasis on the development of mycology through various activities associated with mycological education, training, research, and service in countries and regions within Asia.
As one of the sponsors of the congress, Pensoft proudly presented a Best Talk award to Dr Sinang Hongsanan of Chiang Mai University, Thailand. The award entitles the winner to a free publication in Pensoft’s flagship mycology journal, MycoKeys.
Joint ESENIAS and DIAS Scientific Conference 2023
The ESENIAS and DIAS conference took place from 11-14 October and focused on “globalisation and invasive alien species in the Black Sea and Mediterranean regions.” Pensoft shared information on their NeoBiota journal and the important REST-COAST and B-Cubed projects.

Polina Nikova of the Bulgarian Academy of Sciences received the NeoBiota Best Talk Award for her presentation titled “First documented records in the wild of American mink (Neogale vision von Schreber, 1776) in Bulgaria.” The award entitles her to a free publication in the NeoBiota journal.
XII European Congress of Entomology
Pensoft took part in the XII European Congress of Entomology (ECE 2023) in Heraklion, Crete, from 16-20 October. The event provided a forum for entomologists from all over the world, bringing together over 900 scientists from 60 countries.

The ECE 2023, organised by the Hellenic Entomological Society, addressed the pressing challenges facing entomology, including climate change, vector-borne diseases, biodiversity loss, and the need to sustainably feed a growing world population. The program featured symposia, lectures, poster sessions, and other types of activities aimed at fostering innovation in entomology. For Pensoft, they were a great opportunity to interact with scientists and share their commitment to advancing entomological research and addressing the critical challenges in the field.
Throughout the event, conference participants could find Pensoft’s team at thir booth, and learn more about the scholarly publisher’s open-access journals in entomology. In addition, the Pensoft team presented the latest outcomes from the Horizon 2020 projects B-GOOD, Safeguard, and PoshBee, where the publisher takes care of science communication and dissemination as a partner.
XIV International Congress of Orthopterology 2023
The XIV International Congress of Orthopterology, held from 16-19 October in Mérida, Yucatán, México, was a landmark event in the field of orthopterology.

Hosted for the first time in Mexico, it attracted experts and enthusiasts from around the world. The congress featured plenary speakers who presented cutting-edge research and insights on various aspects of grasshoppers, crickets, and related insects.
Pensoft’s Journal of Orthoptera Research was represented by Tony Robillard, the editor-in-chief, who presented the latest developments of the journal to attendees.
Symposia, workshops, and meetings facilitated discussions on topics like climate change impacts, conservation, and management of Orthoptera. The event also included introductions to new digital and geospatial tools for Orthoptera research.
The 16th International Conference on Ecology and Management of Alien Plant Invasions
The 16th International Conference on Ecology and Management of Alien Plant Invasions (EMAPI 2023) took place in Pucón, Chile, from 23-25 October . The conference focused on the promotion of diversity in the science and management of biological invasions. Several NeoBiota authors ran sessions at the conference, and the journal also presented a Best Talk Award.

4th International ESP Latin America and Caribbean Conference
The 4th International ESP Latin America and Caribbean Conference (ESP LAC 2023) was held in La Serena, Chile, from 6-10 November. Focused on “Sharing knowledge about ecosystem services and natural capital to build a sustainable future,” the event attracted experts in ecosystem services, particularly from Latin America and the Caribbean.
Organised by the Ecosystem Services Partnership, this bi-annual conference was open to both ESP members and non-members, featuring a hybrid format in English and Spanish. Attendees enjoyed an excursion to La Serena’s historical center, adding a cultural dimension to the event.
The conference included diverse sessions and a special recognition by Pensoft’s One Ecosystem journal, which awarded full waivers for publication to the authors of the three best posters.

Magaly Aldave of the Transdisciplinary Center for FES-Systemic Studies claimed first prize with “The voice of children in the conservation of the urban wetland and Ramsar Site Pantanos de Villa in Metropolitan Lima, Peru.” Ana Catalina Copier Guerrero and Gabriela Mallea-Rebolledo, both of the University of Chile, were awarded second and third prize respectively.
Biosystematics 2023
Biosystematics 2023, held from 26-30 November at the Australian National University in Canberra, was a collaborative effort of the Australian Biological Resources Study, Society of Australian Systematic Biologists, Australasian Mycological Society, and Australasian Systematic Botany Society. Themed “Celebrating the Past | Planning the Future,” the conference provided a platform for exploring advancements in biosystematics.
The event featured in-person and online participation, catering to a wide audience of researchers, academics, and students. It included workshops, presentations, and discussions, with a focus on enhancing understanding in biosystematics.
Pensoft awarded three student prizes at the event. Putter Tiatragu, Australian National University, received the Best Student Talk award and a free publication in any Pensoft journal for “A big burst of blindsnakes: Phylogenomics and historical biogeography of Australia’s most species-rich snake genus.”
Helen Armstrong, Murdoch University, received the Best Student Lightning Talk for “An enigmatic snapper parasite (Trematoda: Cryptogonimidae) found in an unexpected host.” Patricia Chan, University of Wisconsin-Madison, was the Best Student Lightning Talk runner-up for “Drivers of Diversity of Darwinia’s Common Scents and Inflorescences with Style: Phylogenomics, Pollination Biology, and Floral Chemical Ecology of Western Australian Darwinia (Myrtaceae).”
–
As we approach the end of 2023, Pensoft looks back on its most prolific and meaningful year of conferences and events. Thank you to everyone who contributed to or engaged with Pensoft’s open-access journals, and here’s to another year of attending events, rewarding important research, and connecting with the scientific community.
***
Follow Pensoft on social media: